ABSTRACT
Glucose transporter type 1 (GLUT1) deficiency syndrome (DS) is a metabolic brain disorder caused by a deficiency resulting from SLC2A1 gene mutation and is characterized by abnormal brain metabolism and associated metabolic encephalopathy. Reduced glucose supply to the brain leads to brain damage, resulting in delayed neurodevelopment in infancy and symptoms such as eye abnormalities, microcephaly, ataxia, and rigidity. Treatment options for GLUT1 DS include ketogenic diet (KD), pharmacotherapy, and rehabilitation therapy. Of these, KD is an essential and the most important treatment method as it promotes brain neurodevelopment by generating ketone bodies to produce energy. This case is a focused study on intensive KD nutritional intervention for an infant diagnosed with GLUT1 DS at Gangnam Severance Hospital from May 2022 to January 2023. During the initial hospitalization, nutritional intervention was performed to address poor intake via the use of concentrated formula and an attempt was made to introduce complementary feeding. After the second hospitalization and diagnosis of GLUT1 DS, positive effects on the infant’s growth and development, nutritional status, and seizure control were achieved with minimal side effects by implementing KD nutritional intervention and adjusting the type and dosage of anticonvulsant medications. In conclusion, for patients with GLUT1 DS, it is important to implement a KD with an appropriate ratio of ketogenic to nonketogenic components to supply adequate energy. Furthermore, individualized and intensive nutritional management is necessary to improve growth, development, and nutritional status.
-
Keywords: Glut1 deficiency syndrome; Diet, ketogenic; Diet, carbohydrate-restricted; Seizures; Epilepsy
INTRODUCTION
Glucose transporters (GLUTs) are involved in transporting glucose to various organs, such as the brain, stomach, muscle, liver, pancreas, and kidneys [
1].
Of the GLUT family members, GLUT1, encoded by the SLC2A1 gene, exhibits substrate specificity and is found in cells of all mammals, predominantly in red blood cells. GLUT1 is involved in metabolism in the brain, facilitating the entry of glucose into the organ [
1,
2].
The glucose requirement of the brain in children is 3–4 times higher than that in adults, accounting for up to 80% of the total systemic glucose utilization, highlighting the significant impact of glucose supply to brain development [
3].
GLUT1 deficiency syndrome (DS) is a disease caused by a mutation in the SLC2A1 gene, resulting in a reduced supply of glucose to the brain and subsequent brain damage. Symptoms include delayed neurodevelopment during infancy, ocular abnormalities, microcephaly, motor dysfunction, and stiffness. The diagnosis is confirmed based on these clinical signs as well as with lumbar puncture showing hypoglycorrhachia and genetic analysis showing pathogenic SLC2A1 variants.
As glucose transport to the brain is impaired, it is essential to secure energy for brain neurodevelopment via ketone bodies as glucose alone cannot provide sufficient energy to the central nervous system [
4].
In GLUT1 DS, KD is an essential and the most important treatment method [
5]. Early implementation of KD has shown effectiveness in improving seizure symptoms, receiving significant clinical attention [
6]. KD comprises a high-fat, low-carbohydrate, and adequate protein meal and has been used for decades in the treatment of epilepsy. Ketones, e.g., β-hydroxybutyrate and acetoacetate, are produced when stored body fats are broken down during fasting. Ketones are involved in energy metabolism, especially in the brain. KD promotes ketone production, maintaining a metabolic state similar to that of fasting [
7].
Currently, there are 4 types of KD: classic KD, modified Atkins diet (MAD), medium chain triglyceride diet, and low glycemic index treatment [
4,
5]. Gastrointestinal disorders, such as constipation, vomiting, and abdominal pain, are the most common side effects of long-term KD. Other known side effects include dyslipidemia, kidney stones, hypoglycemia, metabolic acidosis, iron deficiency anemia, and hypomagnesemia [
8].
This is a report of the growth and development, nutritional status, and clinical effects of intensive nutritional intervention in a pediatric patient diagnosed with GLUT1 DS requiring KD. This study was approved by the Institutional Review Board (IRB) (3-2022-0135) of Gangnam Severance Hospital.
CASE
The patient was born at intrauterine pregnancy 33 weeks, weighing 2.46 kg, via spontaneous vaginal delivery. At birth, the infant had a strong amniotic fluid odor and was suspected of having respiratory distress syndrome, requiring ventilator care for 2 weeks before discharge. Five months later, following a seizure lasting 15 minutes, the infant was admitted to another hospital for treatment but maintained normal development without seizures. At 11 months of age, the infant was admitted to the hospital because of high fever and seizures, exhibited severe refusal of solid foods with persistent seizure symptoms and poor appetite, and was unable to sit or hold himself up.
The infant was taking vitamin B complex, iron supplements, carnitine supplements (L-Carn sol), and an anticonvulsant (Orfil syrup).
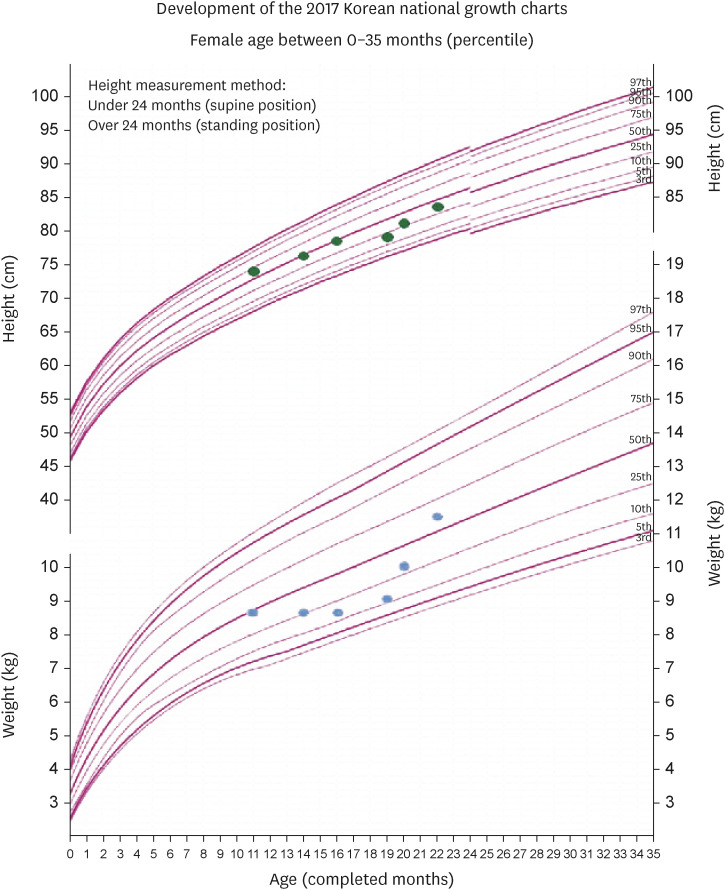
Table 1 and
Figure 1 summarize the results of anthropometric measurements during the nutritional intervention period.
Table 1 Changes of anthropometric data
Table 1
|
Variables |
After birth (mon) |
|
11 |
14 |
16 |
19 |
20 |
22 |
|
Height (cm) |
74.7 |
76.8 |
78.0 |
79.8 |
81.2 |
83.9 |
|
Weight (kg) |
8.8 |
8.8 |
8.8 |
9.1 |
10.1 |
11.5 |
|
Height for age (%ile) |
77.8 |
56.2 |
41.4 |
25.8 |
30.9 |
30.5 |
|
Weight for age (%ile) |
53.0 |
29.8 |
18.5 |
12.8 |
33.3 |
56.5 |
|
Z-score (weight for height) |
−0.3 |
−0.7 |
−1.1 |
−1.0 |
−0.2 |
0.5 |
Figure 1 Changes in nutritional status based on height, weight-for-age.
Fourteen months old (May 2022)
Initial nutritional consultation was requested owing to overall developmental delay observed in the pediatric neurological development assessment.
The weight-for-height z-score was −0.7, which indicated that the patient had a normal nutritional status based on the criteria suggested by the American Society of Parenteral and Enteral Nutrition in 2014 [
9]. The energy intake was 51%, and protein intake was 48% of the nutritional requirements. Nutritional intervention was performed by a dietitian to increase the energy intake using concentrated formula and nutritional supplement drinks, and attempts were made to introduce complementary feeding.
The infant continued taking vitamin B complex and iron supplements, and the anticonvulsant was changed to Keppra solution 70 mg twice daily.
Sixteen months old (July 2022)
The infant was diagnosed with GLUT1 DS via genetic testing, and KD was started.
The weight-for-height z-score was −1.1, indicating mild malnutrition. The patient continued to consume infant formula; however, the energy and protein intake remained insufficient at 55% and 60% of the nutritional requirements, respectively.
An attempt was made to introduce complementary feeding and continue with the formula at a MAD ratio of 1.3:1, but severe refusal of solid foods led to discontinuation of the attempt. The infant underwent nasogastric tube insertion and was transitioned to a KD ratio of 4:1. Energy intake was limited to 75%–85% of nutritional requirements [
10,
11], providing a total energy of 864 kcal, 7.2 g of carbohydrates, 14.4 g of protein, and 86.4 g of fat, supplied via ketogenic products 4 times a day. KD was initiated without fasting to prevent hypoglycemic side effects [
5,
8,
11]. To adapt to KD, one-third of the nutritional supply was provided on the first day, two-thirds on the second day, and full supply from the third day onward, with education to avoid consuming foods other than KD. However, oral intake was resumed only after removing the nasogastric tube, because of the patient’s refusal to eat with the tube. After 3 months, a gastrostomy tube was placed and attempts to bring the KD ratio to 4:1 were resumed.
Appetite stimulants, vitamin B complex, and the existing anticonvulsant (Keppra sol 82.5 mg twice daily) were increased, and Trileptal suspension 60 mg twice daily was added.
Nineteen months old (October 2022)
The infant had a weight-for-height z-score of −1.0, indicating mild malnutrition while consuming formula in proportion to the nutritional requirements at 66% for energy and 80% for protein. After gastrostomy, the total energy of KD was incrementally increased to 648, 720, and 864 kcal at a ratio 4:1. However, the infant subsequently experienced more severe gastrointestinal issues, including vomiting and diarrhea. Considering that the diarrhea persisted even after 1 week, the KD ratio was changed to 3:1.
Protein powder was added to the ketogenic products to adjust KD to 3:1, with a total energy of 889 kcal, 7.2 g of carbohydrates, 21.6 g of protein, and 86.4 g of fat.
Eleven days after discharge, despite consuming the diet at 100%, the infant experienced constipation and the caregiver reported that formula preparation was inconvenient, leading to an adjustment back to KD 4:1, with a total energy of 864 kcal via a phone consultation.
Vitamin B complex, iron supplements, and the existing anticonvulsant Keppra sol 82.5 mg twice daily were continued.
Twenty months old (November 2022)
KD was adjusted to a ratio of 4:1, with a total energy of 864 kcal, maintaining 100% intake. With the use of this ratio, there were no issues related to diarrhea or gastrointestinal problems, but there was a persistent decline in the overall condition. Thus, KD was readjusted to a ratio of 3:1, with a total energy of 889 kcal.
The weight-for-height z-score was −0.2, indicating no malnutrition. The infant showed increased interest in the tools and their manipulation as well as more emotional expression.
However, the pediatric neurological development assessment showed a significant delay compared with children of the same age. Independent walking attempts did not appear, and active therapy for gross motor development was reported as necessary.
Medications included vitamin B complex, carnitine supplement (L-Carn sol), a reduced dose of the existing anticonvulsant Keppra solution (62.5 mg twice daily), and the addition of a new anticonvulsant, Promag (66–100–150 mg twice daily).
Twenty-two months old (January 2023)
KD was maintained for 3 months at a ratio of 3:1, with a total energy intake of 889 kcal, and the weight-for-height z-score was 0.5, indicating no malnutrition.
The infant’s overall condition improved, and there was a reduction in seizures, with the electroencephalograms reporting only mild diffuse encephalopathy. The infant was able to stand independently and had stools of Bristol stool chart type 4. Attempts were made to introduce mashed foods such as soft tofu, eggs, and cheese for oral consumption in small amounts via nutritional education.
The medications included vitamin B complex, carnitine supplement (L-Carn sol), and Promag (200 mg twice daily).
Table 2 summarizes the clinical nutritional management of the patient with GLUT1 DS.
Table 2Summary of nutrition intervention
Table 2
|
Variables |
After birth (mon) |
|
11 |
14 |
16 |
19 |
20 |
22 |
|
The contents of nutrition intervention |
- |
Failure to thrive |
Modified Atkins diet → Ketogenic diet |
Ketogenic diet |
Ketogenic diet |
Ketogenic diet |
|
The daily energy requirements (kcal) |
- |
700 |
1,000 |
1,100 |
1,100 |
1,100 |
|
The daily energy supply (kcal) |
- |
700 |
864 |
880 |
880 |
880 |
|
The daily energy intake (kcal) |
- |
373 |
440 → 0 |
889 |
889 |
889 |
|
the daily carbohydrate supply (g) |
- |
- |
12.0 → 7.2 |
7.2 |
7.2 |
7.2 |
|
The daily protein supply (g) |
- |
25.0 |
39 → 14.4 |
21.6 |
21.6 |
21.6 |
|
The daily fat supply (g) |
- |
- |
67 → 86.4 |
86.4 |
86.4 |
86.4 |
|
The ratio of fat to carbohydrate & protein |
- |
- |
1.3:1 → 4:1 |
4:1 → 3:1 → 4:1 |
4:1 → 3:1 |
3:1 |
|
The feeding method |
- |
Oral |
Nasogastric tube |
Gastrostomy tube |
Gastrostomy tube |
Gastrostomy tube |
|
The problems of eating |
Anorexia |
Anorexia, oral feeding refuse |
Tube feeding refuse |
Gastrointestinal symptom, diarrhea |
Constipation, drool |
Drool |
Lab results
From 19 months of age, when KD was initiated, the urine ketone levels increased and remained between +2 and +3.
The patient’s lipid level increased at the start of KD and then showed a decreasing trend over time. No abnormalities such as hypoglycemia, hypomagnesemia were present; however, mild metabolic acidosis was observed.
Table 3 summarizes the biochemical test results of the infant with GLUT1 DS.
Table 3Changes of biochemical data
Table 3
|
Variables |
After birth (mon) |
|
11 |
14 |
16 |
19 |
20 |
22 |
|
Glucose (mg/dL) |
- |
80.0 |
99.0 |
81.0 |
71.0 |
78.0 |
|
BUN (mg/dL) |
- |
9.30 |
14.70 |
10.90 |
6.10 |
9.80 |
|
Creatinine (mg/dL) |
- |
0.21 |
0.19 |
0.29 |
0.15 |
0.13 |
|
Cholesterol (mg/dL) |
- |
168.0 |
160.0 |
93.0 |
107.0 |
135.0 |
|
Triglyceride (mg/dL) |
- |
- |
71.0 |
177.0 |
128.0 |
425.0 |
|
HDL-cholesterol (mg/dL) |
- |
- |
42.0 |
29.0 |
34.0 |
30.0 |
|
LDL-cholesterol (mg/dL) |
- |
- |
103.8 |
28.6 |
47.4 |
74.0 |
|
Magnesium (mmol/L) |
- |
- |
0.88 |
0.82 |
0.80 |
0.72 |
|
tCO2 (mmol/L) |
- |
23.0 |
22.0 |
18.0 |
19.0 |
21.0 |
|
pH |
- |
- |
7.458 |
7.469 |
7.391 |
- |
|
HCO3- (mmol/L) |
- |
- |
19.6 |
18.5 |
17.5 |
- |
|
Urine ketone |
- |
- |
- |
2+ |
3+ |
- |
DISCUSSION
This case report reported the nutritional management effects of implementing KD after the diagnosis of GLUT1 DS.
Previous studies reported that applying KD positively affect the symptoms of GLUT1 DS [
4,
7,
12,
13,
14]. Currently, KD is considered as the gold standard treatment for GLUT1 DS as it increases the level of ketone bodies in the blood for use by the brain. Considering that children need more energy for their brains to grow, early diagnosis of GLUT1 DS and the initiation and continuation of KD at least until adolescence, is crucial [
15].
In this case, KD was initiated via gastrostomy for cranial nerve development in a GLUT1 DS infant who refused oral intake. Although the classic KD is usually implemented after a period of fasting, recent studies have reported that starting KD without fasting results in higher compliance, lower risk of dehydration and hypoglycemia, and no significant difference in the anticonvulsant effect [
5]. Therefore, in this case, the patient was directly provided KD without the initial fasting, which facilitated adaptation.
To improve compliance, ongoing nutritional interventions were performed, including adjustment of the KD ratio and nutrition intake.
The weight-for-height z-score (before KD –0.3, after 0.5), which is an important measure of nutritional status in children, increased, which indicates improved nutritional status. KD 3:1 customized for the infant reduced the side effects such as seizures and gastrointestinal problems. Furthermore, there were no abnormalities observed in the biochemical test results, including initial increase of the lipid profile, plasma glucose, and magnesium concentrations. These positive effects demonstrate the importance of intensive nutritional management via KD. However, there is a need for active rehabilitation therapy and ongoing management owing to motor impairment and cognitive development being below the expected level for the age.
In particular, it should be noted that KD is a high-fat diet that causes dyslipidemia due to potentially increased triglyceride, total cholesterol, and low-density lipoprotein-cholesterol. The multidisciplinary team should control the KD rate and minimize the risk of cardiovascular disease through continuous monitoring.
To improve the effectiveness of KD and achieve the treatment goals, multidisciplinary care comprising doctors, nurses, and dietitians as well as correct dietary provision, education, and practice by patients and their caregivers are critical. The doctor prescribes the rate of KD according to the patient’s condition and observes the clinical condition. The nurse confirms ketone adaptation and provides instructions for emergency management. The dietitian provides the appropriate diet plan according to the KD ratio and educates the caregiver on how to calculate and adjust the diet to meet the KD ratio to ensure that it is practiced after discharge and monitored continuously. Both healthcare professionals and caregivers need to have a positive attitude toward the dietary intervention, understanding that KD is a therapeutic method rather than just a meal. Maintaining KD is vital for the quality of life of the patient and caregivers as the condition requires lifelong KD treatment.
A limitation of this case report is that medications were frequently changed to control seizures.
Whether the effects of seizure control were due solely to dietary therapy is not clear. Further research on the clinical effectiveness of KD adjustment in patients with GLUT1 DS is needed.
For the above patient, the feeding was done via a feeding tube using products with a balanced ketogenic to nonketogenic ratio, which was easy to maintain and manage. However, when consumed as a food, it is considered that it would be difficult to maintain the ketogenic to nonketogenic ratio, adjust the nutrient content, and manage it over the long term. GLUT1 DS is a condition that requires lifelong treatment with KD, thereby affecting the quality of life of the patients and their caregivers.
In conclusion, KD is essential for the treatment of patients with GLUT1 DS and maintaining an appropriate ketogenic to nonketogenic ratio would ensure an adequate energy supply. While it may take several months for the effects to show, early diagnosis and intensive nutritional intervention can have a positive impact on growth and development, nutritional status, and clinical outcomes.
NOTES
-
Conflict of Interest: The authors declare that they have no competing interests.
-
Author Contributions:
Conceptualization: Kim YS, Kim W, Na JH, Lee YM.
Writing - original draft: Kim YS.
Writing - review & editing: Kim YS, Na JH.
REFERENCES
- 1. Koch H, Weber YG. The glucose transporter type 1 (Glut1) syndromes. Epilepsy Behav 2019;91:90-93.
- 2. Gumus H, Bayram AK, Kardas F, Canpolat M, Çağlayan AO, Kumandas S, Kendirci M, Per H. The effects of ketogenic diet on seizures, cognitive functions, and other neurological disorders in classical phenotype of glucose transporter 1 deficiency syndrome. Neuropediatrics 2015;46:313-320.
- 3. Klepper J, Akman C, Armeno M, Auvin S, Cervenka M, Cross HJ, De Giorgis V, Della Marina A, Engelstad K, Heussinger N, Kossoff EH, Leen WG, Leiendecker B, Monani UR, Oguni H, Neal E, Pascual JM, Pearson TS, Pons R, Scheffer IE, Veggiotti P, Willemsen M, Zuberi SM, De Vivo DC. Glut1 deficiency syndrome (Glut1DS): state of the art in 2020 and recommendations of the international Glut1DS study group. Epilepsia Open 2020;5:354-365.
- 4. Schwantje M, Verhagen LM, van Hasselt PM, Fuchs SA. Glucose transporter type 1 deficiency syndrome and the ketogenic diet. J Inherit Metab Dis 2020;43:216-222.
- 5. Kossoff EH, Zupec-Kania BA, Auvin S, Ballaban-Gil KR, Christina Bergqvist AG, Blackford R, Buchhalter JR, Caraballo RH, Cross JH, Dahlin MG, Donner EJ, Guzel O, Jehle RS, Klepper J, Kang HC, Lambrechts DA, Liu YM, Nathan JK, Nordli DR Jr, Pfeifer HH, Rho JM, Scheffer IE, Sharma S, Stafstrom CE, Thiele EA, Turner Z, Vaccarezza MM, van der Louw EJ, Veggiotti P, Wheless JW, Wirrell EC. Charlie Foundation. Matthew’s Friends. Practice Committee of the Child Neurology Society. Optimal clinical management of children receiving dietary therapies for epilepsy: updated recommendations of the International Ketogenic Diet Study Group. Epilepsia Open 2018;3:175-192.
- 6. Kass HR, Winesett SP, Bessone SK, Turner Z, Kossoff EH. Use of dietary therapies amongst patients with GLUT1 deficiency syndrome. Seizure 2016;35:83-87.
- 7. Klepper J, Diefenbach S, Kohlschütter A, Voit T. Effects of the ketogenic diet in the glucose transporter 1 deficiency syndrome. Prostaglandins Leukot Essent Fatty Acids 2004;70:321-327.
- 8. Kang HC, Chung DE, Kim DW, Kim HD. Early- and late-onset complications of the ketogenic diet for intractable epilepsy. Epilepsia 2004;45:1116-1123.
- 9. Becker PJ, Nieman Carney L, Corkins MR, Monczka J, Smith E, Smith SE, Spear BA, White JV. Consensus statement of the Academy of Nutrition and Dietetics/American Society for Parenteral and Enteral Nutrition: indicators recommended for the identification and documentation of pediatric malnutrition (undernutrition). J Acad Nutr Diet 2014;114:1988-2000.
- 10. Sampaio LP. Ketogenic diet for epilepsy treatment. Arq Neuropsiquiatr 2016;74:842-848.
- 11. Severance Hospital. Samsung Welstory. Ketogenic eating guide. Georgia: Cypress; 2016.
- 12. Veggiotti P, Teutonico F, Alfei E, Nardocci N, Zorzi G, Tagliabue A, De Giorgis V, Balottin U. Glucose transporter type 1 deficiency: ketogenic diet in three patients with atypical phenotype. Brain Dev 2010;32:404-408.
- 13. Ramm-Pettersen A, Stabell KE, Nakken KO, Selmer KK. Does ketogenic diet improve cognitive function in patients with GLUT1-DS? A 6- to 17-month follow-up study. Epilepsy Behav 2014;39:111-115.
- 14. Ramm-Pettersen A, Nakken KO, Skogseid IM, Randby H, Skei EB, Bindoff LA, Selmer KK. Good outcome in patients with early dietary treatment of GLUT-1 deficiency syndrome: results from a retrospective Norwegian study. Dev Med Child Neurol 2013;55:440-447.
- 15. Olivotto S, Duse A, Bova SM, Leonardi V, Biganzoli E, Milanese A, Cereda C, Bertoli S, Previtali R, Veggiotti P. Glut1 deficiency syndrome throughout life: clinical phenotypes, intelligence, life achievements and quality of life in familial cases. Orphanet J Rare Dis 2022;17:365.